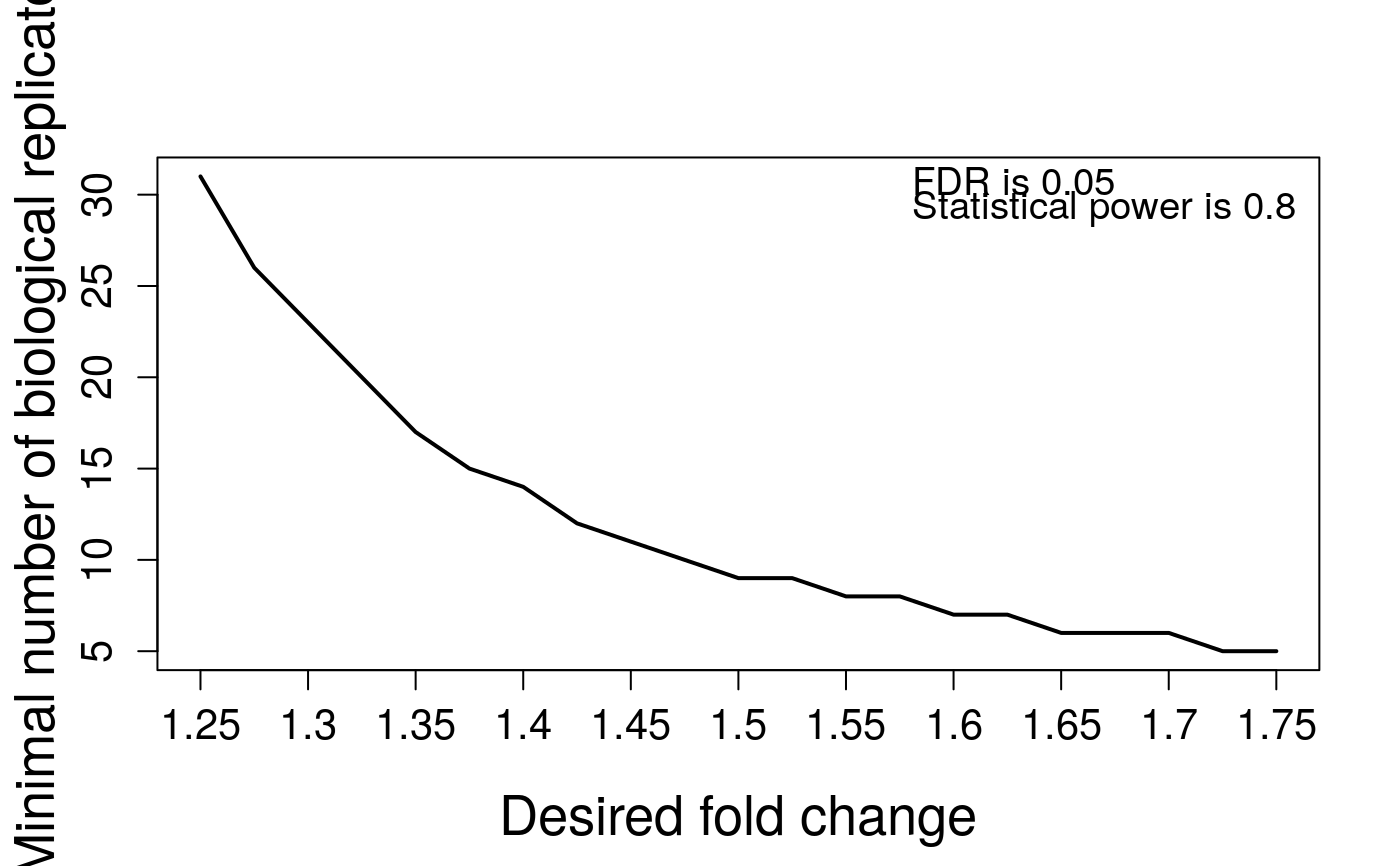
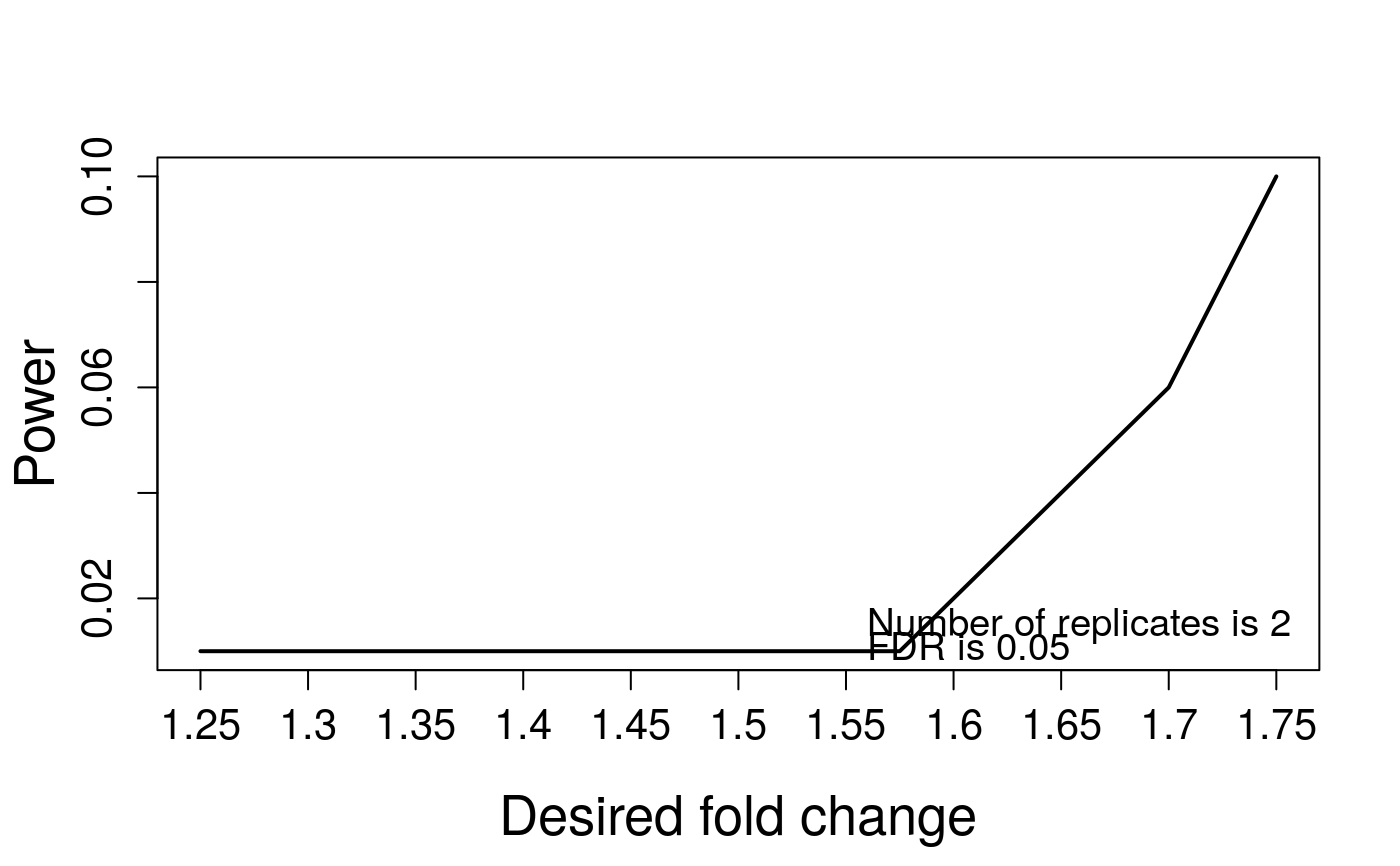
Visualization for sample size calculation
designSampleSizePlots.RdTo illustrate the relationship of desired fold change and the calculated
minimal number sample size which are (1) number of biological replicates per condition,
(2) number of peptides per protein,
(3) number of transitions per peptide, and
(4) power. The input is the result from function (designSampleSize.
designSampleSizePlots(data)
Arguments
| data | output from function designSampleSize. |
|---|
Value
Plot for estimated sample size with assigned variable.
Details
Data in the example is based on the results of sample size calculation from function designSampleSize
Examples
# Based on the results of sample size calculation from function designSampleSize, # we generate a series of sample size plots for number of biological replicates, or peptides, # or transitions or power plot. QuantData<-dataProcess(SRMRawData)#> INFO [2021-07-05 20:05:41] ** Features with one or two measurements across runs are removed. #> INFO [2021-07-05 20:05:41] ** Fractionation handled. #> INFO [2021-07-05 20:05:41] ** Updated quantification data to make balanced design. Missing values are marked by NA #> INFO [2021-07-05 20:05:41] ** Log2 intensities under cutoff = 3.776 were considered as censored missing values. #> INFO [2021-07-05 20:05:41] ** Log2 intensities = NA were considered as censored missing values. #> INFO [2021-07-05 20:05:41] ** Use all features that the dataset originally has. #> INFO [2021-07-05 20:05:41] #> # proteins: 2 #> # peptides per protein: 2-2 #> # features per peptide: 3-3 #> INFO [2021-07-05 20:05:41] #> 1 2 3 4 5 6 7 8 9 10 #> # runs 3 3 3 3 3 3 3 3 3 3 #> # bioreplicates 3 3 3 3 3 3 3 3 3 3 #> # tech. replicates 1 1 1 1 1 1 1 1 1 1 #> INFO [2021-07-05 20:05:41] == Start the summarization per subplot... #> | | | 0% | |=================================== | 50% | |======================================================================| 100% #> INFO [2021-07-05 20:05:42] == Summarization is done.#> NULL## based on multiple comparisons (T1 vs T3; T1 vs T7; T1 vs T9) comparison1<-matrix(c(-1,0,1,0,0,0,0,0,0,0),nrow=1) comparison2<-matrix(c(-1,0,0,0,0,0,1,0,0,0),nrow=1) comparison3<-matrix(c(-1,0,0,0,0,0,0,0,1,0),nrow=1) comparison<-rbind(comparison1,comparison2, comparison3) row.names(comparison)<-c("T3-T1","T7-T1","T9-T1") colnames(comparison)<-unique(QuantData$ProteinLevelData$GROUP) testResultMultiComparisons<-groupComparison(contrast.matrix=comparison, data=QuantData)#> INFO [2021-07-05 20:05:42] == Start to test and get inference in whole plot ... #> | | | 0% | |=================================== | 50% | |======================================================================| 100% #> INFO [2021-07-05 20:05:42] == Comparisons for all proteins are done.# plot the calculated sample sizes for future experiments: # (1) Minimal number of biological replicates per condition result.sample<-designSampleSize(data=testResultMultiComparisons$FittedModel, numSample=TRUE, desiredFC=c(1.25,1.75), FDR=0.05, power=0.8) designSampleSizePlots(data=result.sample)# (2) Power result.power<-designSampleSize(data=testResultMultiComparisons$FittedModel, numSample=2, desiredFC=c(1.25,1.75), FDR=0.05, power=TRUE) designSampleSizePlots(data=result.power)